Intrinsic Self-healing Polymers
Self-healing is an intrinsic property of all living organisms. It requires no external intervention, meaning wounds and broken bones heal by itself, and, in some organisms, even lost parts of a living body can be replaced. Like living organisms, plastics, elastomers and composites also suffer from damage caused by excessive heat, radiation, stress and/or chemicals. This leads to a loss of useful properties over time due to microscopic and macroscopic damage.
Particulalry sensitive to mechanical / chemical damage and degradation are engineering and high-performance plastics due to their inherent brittleness and plastics with unsaturated bonds and weak linkages in the polymer backbone. Often damage starts with microcracking induced by excessive thermal and/or mechanical stress or by cyclic loads particularly in aggressive atmospheres. These types of damages are difficult to observe and repair with conventional methods since microcracks are often hidden deep in the structure.
The approaches to self-healing can be divided into two broad categories:1 (a) intrinsic self-healing where the polymers themselves heal molecular and macroscopic damage (cracks), and (b) extrinsic self-healing in which a healing agent, pre-embedded in the polymer matrix, is released into voids (cracks) when the polymer matrix is damaged. Self-healing materials can be further classified as autonomic, where repair is triggered by the damage itself and non-autonomic where repair is triggered by an external action such as reduction / oxidation, pH-change, UV / EB irradiation, pressure or heat.
Intrinsic self-healing systems can also be classified based on their underlying type of healing mechanism. The two main categories are physical interaction and chemical bonding. The former can be only applied to soft (thermoplastic) materials such as hydrogels2 or materials that consist of both mobile soft and immobile rigid domains such as thermoplastic/thermosetting semi-interpenetrating polymer networks. The latter often requires heating to mobilize the thermoplastic portion so that the polymers can diffuse through the thermosetting matrix to close cracks and thereby facilitating healing.3 Most chemical healing methods are non-autonomic. In other words, these methods require an external stimulus such as heat or UV radiation to break and reform bonds.
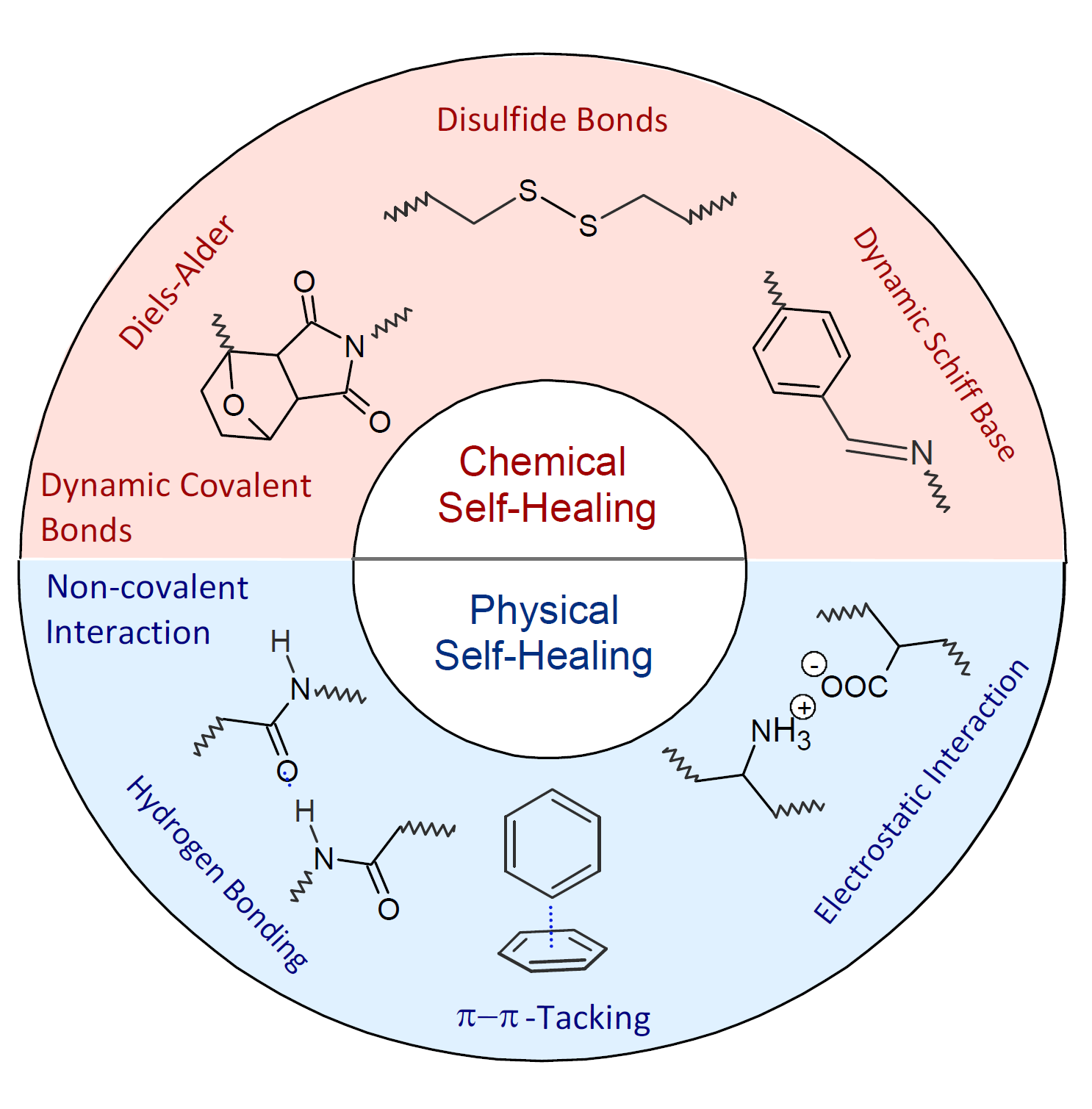
Physical Self-Healing
Physical self-healing is based on non-covalent interactions including diffusion and entanglement, π-π stacking, ionic and hydrophobic interactions, hydrogen bonding, and metal coordination.1
Ionomeric self-healing systems typically consist of thermoplastic ionic copolymers (the so-called ionomers) which are made up of hydrocarbon chains where a portion bears (carboxylic) acid groups that are either partially or completely neutralized with metal or quaternary ammonium ions. A common example is poly(ethylene-co-methacrylic acid) or EMAA. The ionic segments of EMAA can form clusters that act as reversible cross-links which can be broken and reformed by external stimuli such as temperature and UV light. These materials have been studied by Kalista et al.4,5 and Varley & van der Zwaag 6,7
Supramolecular self-healing materials are made up of polymers with strong end-group and/or side-group associations via hydrogen bonding and metal coordination or hydrogen bonding and π-π-stacking.7-9 These rubbery self-healing materials can be fractured and mended by bringing the broken pieces into close contact to allow for reformation of the hydrogen bonds.
Self-healing via molecular diffusion and entanglement is the simplest and probably oldest method of self-healing. Healing of the damaged areas (void closure) occurs via diffusion, entanglement and surface interaction induced by heat and/or pressure. In principle, any thermoplastic material can be healed by this method if the temperature (and pressure) is sufficiently high to allow for diffusion and entanglement of the polymers. For example, O'Connor & Wool11,12 and McGarel & Wool13 showed that this mechanism can be successfully employed to heal damages in styrene-isoprene-styrene block copolymers and in polystyrene.
Chemical Self-Healing
A very popular chemical self-healing mechanism is based on reversible covalent bonding. Compared to physical bonding (supramolecular interactions), chemical reversible bonding methods provide higher mechanical strength and greater dimensional stability. A widely utilized reversible reaction is the [4+2] cycloaddition of a diene to a dienophile which is known as Diels-Alder (DA), whereas the reverse reaction is called retro-Diels-Alder (rDA). The most common diene is furan and the most common dienophile is maleimide. Typically only a portion of the bonds are replaced with these DA bonds. They can be easily broken and reformed when heated allowing the chains to flow into cracks and to form new bonds with the cracked surfaces that leads to a partial recovery of the initial strength. This chemistry has been successfully employed for the development of numerous thermally-induced, self-healable polymers.13-17 Other common dynamic bonds utilized in stimuli responsive self-healing include disulfide bridges,18-20 trithiocarbonates, 21 acylhydrazone bonds,22,23 urea linkages with bulky substituents,24,25 boronic esters,26 and alkoxyamines.27 Some of these dynamic covalent chemistries are shown in the table below.
| Bond Type | Reversible Reaction |
| Diels-Alder | 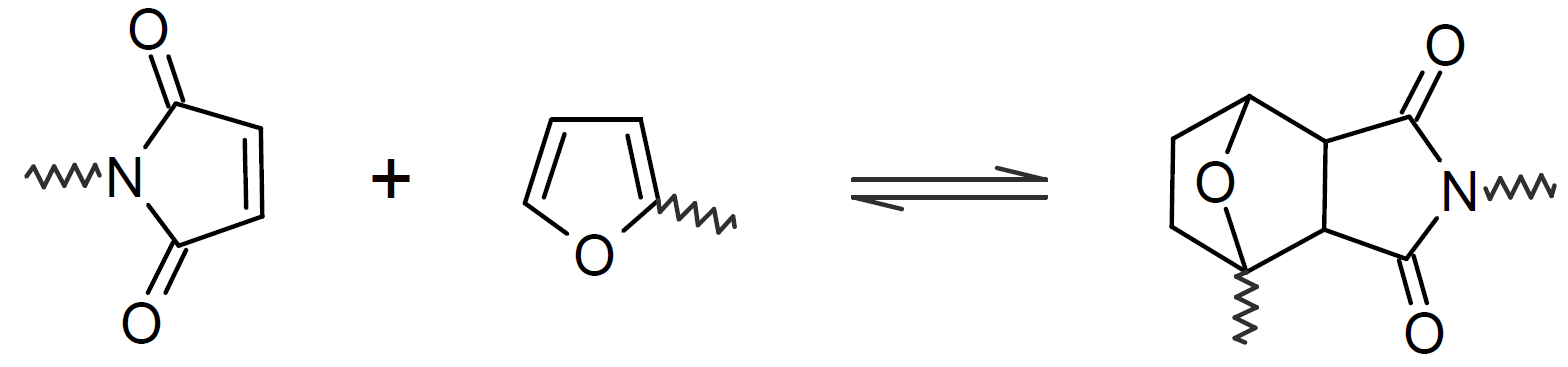 |
| Hindered Urea | 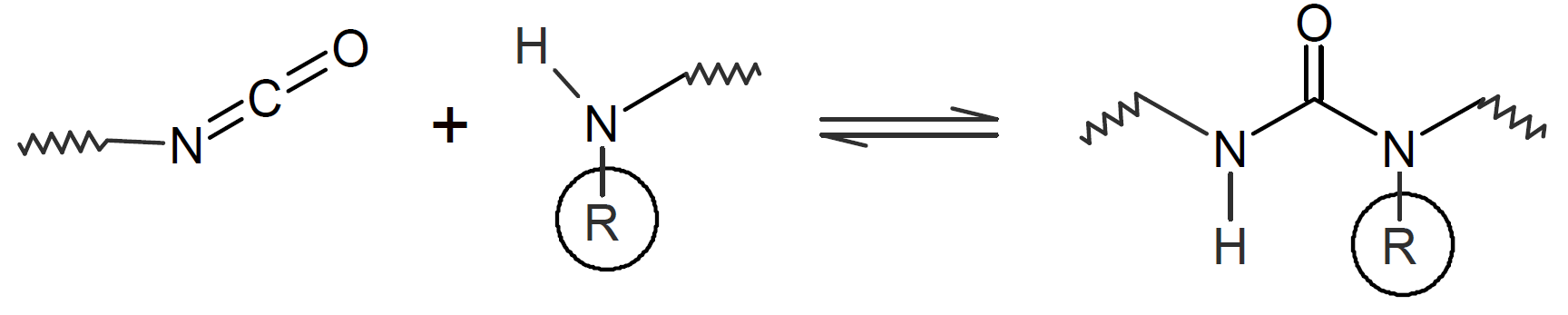 |
| Boronic Ester | 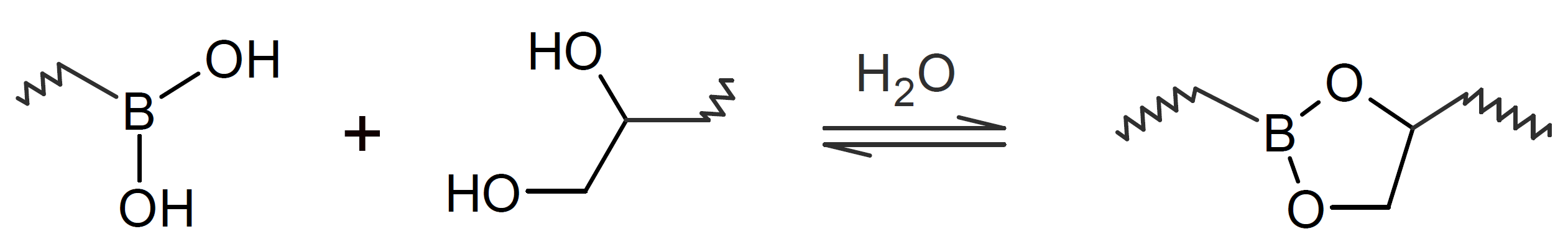 |
| Alkoxy Amine | 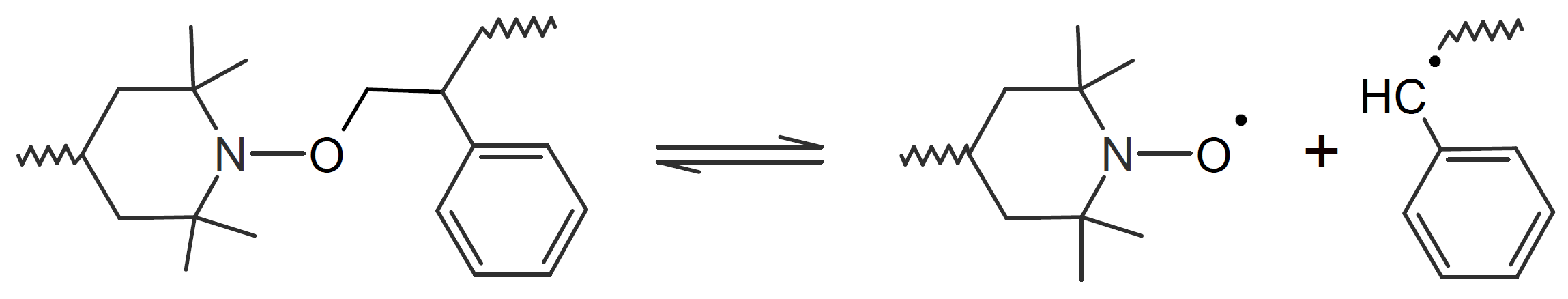 |
| Trithiocarbonate | 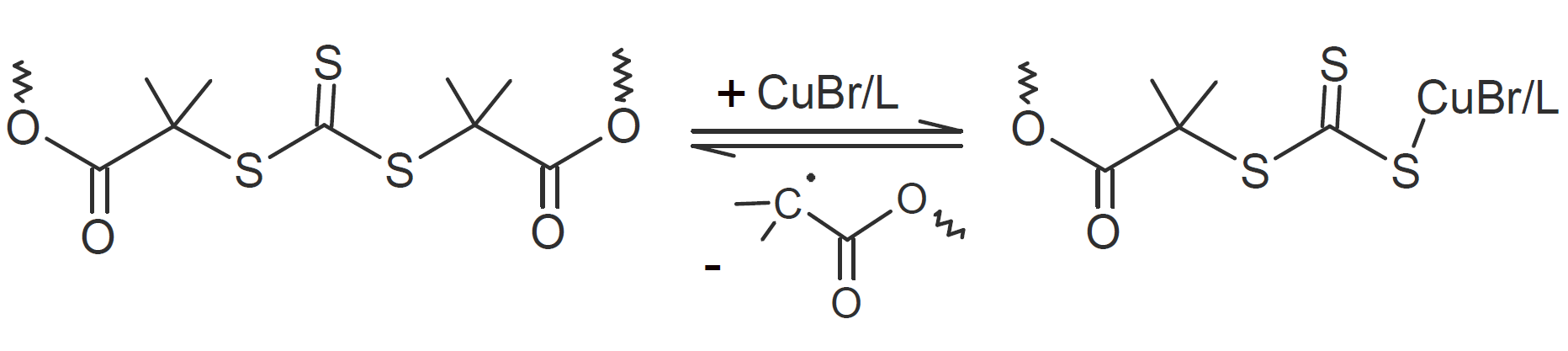 |
| Disulfides | 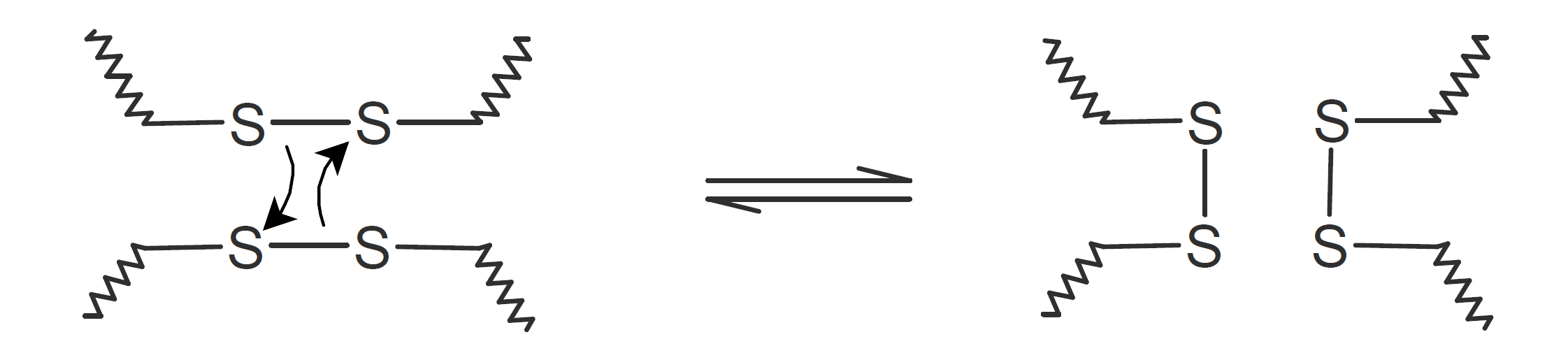 |
| Acylhydrazone | 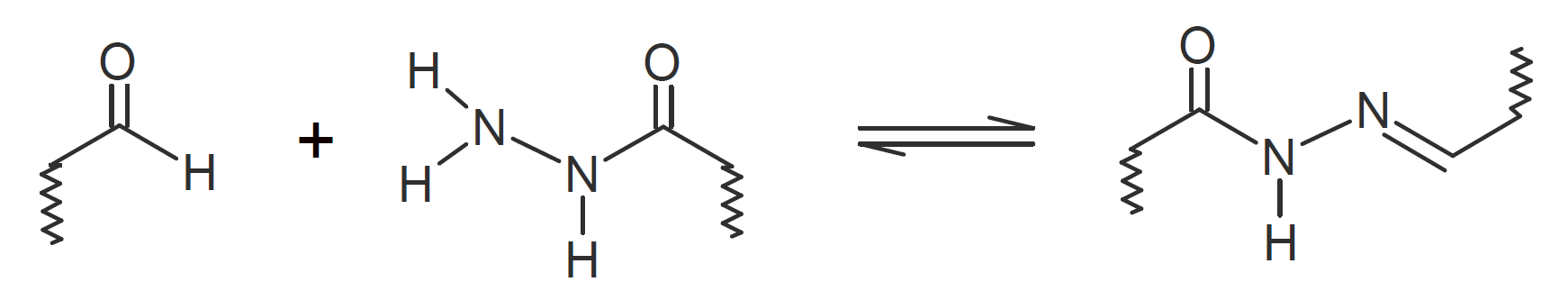 |
It should be noted that many traditional step-growth polymerizations yield polymers with unreacted end groups unless monofunctional monomers are added or the polymerization is driven to very high conversions beyond 99 % where rings will dominate due to intramolecular ring-closure reactions. These types of polymers possess some degree of self-healing, which typically proceeds above the glass transition temperature by interdiffusion of polymers / oligomers into the cracked region where the residual functional groups react with each other leading to a partial recovery of the initial strength.3,28 However, this type of healing is not truly intrinsic since the number of healing events is limited.
References and Notes
B.J. Blaiszik, S.L.B. Kramer, S.C. Olugebefola, J.S. Moore, N.R. Sottos, S.R. White, Annu. Rev. Mater. Res. 40, 179-211 (2010)
P. Michael, D. Doehler, W.H. Binder, Polymer, 69 216-227 (2015)
Y.C. Yuan, T. Yin, M.Z. Rong, M.Q. Zhang, eXPRESS Polym. Lett., Vol. 2, 4, 238-250 (2008)
S.J. Kalista, T.C. Ward, Z. Oyetunji, Mech. Adv. Mater. Struct. 14(5), 391-97 (2007)
S.J. Kalista, T.C. Ward, J. R. Soc. Interface 4(13), 405-11 (2007)
R.J. Varley, S. van der Zwaag, Acta Mater. 56(19), 5737-50 (2008)
R.J. Varley, S. van der Zwaag, Polym. Test. 27(1), 11-19 (2008)
L. Brunsveld, B.J.B. Folmer, E.W. Meijer, R.P. Sijbesma, Chem. Rev. 101(12), 4071-4098 (2001)
K. Chino, and M. Ashiura, Macromolecules, 34(26), 9201-9204 (2001)
S.P. Sanka, B. Krishnakumar, Y. Leterrier, S. Pandey, S. Rana and V. Michaud, Front. in Mat. 6, 137 (2019)
K. O'Connor and R. Wool J. Appl. Phys. 51(10), 5075-79 (1980)
R. Wool and K. O'Connor, J. Appl. Phys. 52(10), 5953-63 (1981)
J.O. McGarel and R.P. Wool, J. Polym. Sci. B 25(12), 2541–60 (1987)
N.I. Khan et al., IOP Conf. Ser.: Mater. Sci. Eng. 377, 012007 (2018)
M.M. Diaz, G. Van Assche, F.H.J. Maurer, B. Van Mele, Polymer 120, 176-188 (2017)
K. Urdl, S. Weiss, P. Christoefl, A. Kandelbauerd, U. Mueller, W. Kern. Eur. Poly. J., Vol 127, 15 (2020)
B. Strachota, J. Hodan, J. Dybal, L. Matejka, Macromol. Mater. Eng. 306(1) (2021)
B. Krishnakumar, M. Singh, V. Parthasarthy, C. Park, N.G. Sahoo, G.J. Yun, S. Rana , Nanoscale Adv. 2, 2726 (2020)
I. Degirmenci, and M.L. Coote, JOTCSA. 3(3), 707-720 (2016)
S. Nevejans, N. Ballard, J.I. Miranda, B. Reck and J.M. Asua, Phys. Chem. Chem. Phys., 18, 27577 (2016)
R. Nicolay, J. Kamada, A. Van Wassen and K. Matyjaszewski, Macromolecules 43, 4355 (2010)
G. Deng, C. Tang, F. Li, H. Jiang and Y. Chen, Macromolecules 43, 1191-1194 (2010)
N. Roy, E. Buhler, J.M. Lehn, Polym. Int. 63(8), 1400-5 (2014)
H. Ying, Y. Zhang and J. Cheng, Nat. Commun. 5(1), 3218 (2014)
S. Zechel, R. Geitner, M. Abend, M. Siegmann et al., NPG Asia Mat. 9(8), 420 (2017)
J.J. Cash, T. Kubo, A.P. Bapat and B.S. Sumerlin, Macromolecules 48, 2098-2106 (2015)
Z.P. Zhang, M.Z. Rong, M.Q. Zhang and C. Yuan, Polym. Chem. 4(17), 4648-4654 (2013)
R.P. Wool, Polymer Interfaces: Structure and Strength, Hanser, Munich (1994)